A Practical Guide to Solid Phase Peptide Synthesis (SPPS)
A 65 page guide on the practical aspects of performing SPPS
What is this guide?
Written through more than 60 years of combined experienced in making peptides, this is a 65 page practical guide on SPPS.
The purpose of this guide is two-fold. First, a brief introduction on the development and most common applications of solid-phase peptide synthesis will enable the user to best apply the two most widely-used synthetic strategies – Boc/Benzyl and Fmoc/tButyl chemistries – to his/her projects. Second, a detailed description of peptide synthesis, cleavage, and purification in the experimental section is given with ‘helpful hints’ so that newcomers to peptide science will have easy access and avoid some of the obstacles which often lead to expensive mistakes and/or poor synthesis yields
Introduction
Solid phase synthesis is a process by which chemical transformations can be carried out on solid support in order to prepare a wide range of synthetic compounds. This idea was first developed by Bruce Merrifield to synthesize polypeptides and earned him the Nobel Prize in 1984. Solid phase chemistry offers many advantages over conventional synthesis in terms of efficiency as well as convenient work-up and purification procedures. In solution phase peptide synthesis, particularly in longer sequences, the repetition of coupling and deprotection cycles can become very labor intensive and require the isolation of all peptide intermediates.
I. A Brief Historical Perspective The chemistry of peptide synthesis – first developed in the early 1900’s by Emil Fischer – arguably marks the birth of organic synthesis as we know it today. Whereas the lion share of organic synthesis continues to be performed by solution-phase methods, i.e., with each independent chemical reaction followed by a purification step and characterization of the resulting synthetic intermediate, two peculiarities of peptide chemistry spurred the development of a more efficient synthetic strategy. In contrast to most total synthesis efforts, the synthesis of peptides – at least until the final deprotection step – is an iterative process, with alpha-amino (alphaN) deprotection and amide couplings performed in succession until the desired full-length target peptide is obtained. In addition, most peptides of biological interest are grossly insoluble in most organic solvents irrespective of side-chain protection tactics. The net result of these two characteristic features of peptides was that the first sixty-odd years of peptide synthesis forged little ground until the landmark work in the late 1950’s of R.B. Merrifield at Rockefeller University.
During the course of his Ph.D. work, Merrifield (a biochemist) proposed an entirely new paradigm in organic synthesis. As is often the case when an outsider looks into an insular field and pursues a tack that is anathema to the existing experts in the field, Merrifield’s idea of performing all synthetic manipulations using the C-terminus of the target peptide linked to an insoluble solid support was met with much criticism. However, it was not long before the chemistry of solid-phase peptide synthesis (SPPS) was honed to a point where traditional solution-phase methodologies were no match with regard to speed and versatility. The original “Merrifield” version of SPPS – more accurately referred to as Boc/Benzyl chemistry – was roughly finalized in the late 1960s, and employs a graduated acid lability system for manipulation of all protecting groups (Scheme 1). In this strategy, the alpha-amino t-butoxycarbonyl (Boc) protection is removed with TFA, while side-chain protections and the peptide- resin anchorage (the linker) require much harsher acidic conditions for cleavage. This final step is accomplished using liquid HF, a much stronger acid than TFA (acidity functions of -11 and 0.1, respectively).
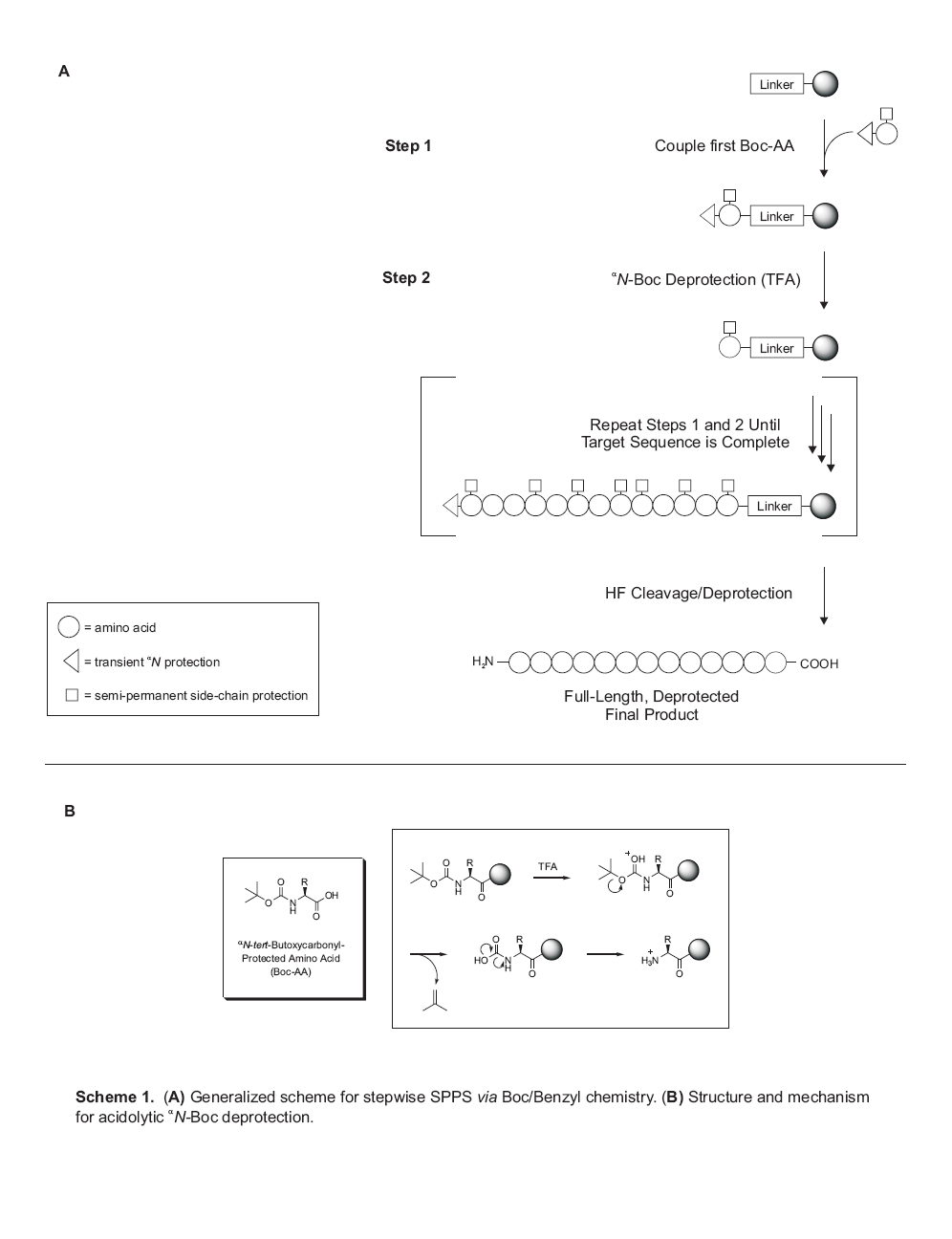
Scheme 1 depicts the manner in which Boc/Benzyl SPPS simplifies all of the reactions involved in peptide synthesis by allowing for purification via filtration, so that excess reagents can be employed and removed by simple washing. It is important to emphasize that the chemistry of SPPS is not fundamentally different from that used in solution-phase peptide synthesis. The sole chemical distinction between solution- and solid-phase peptide synthesis is that the C-terminal protecting group in the latter is rendered insoluble by virtue of its incorporation into a polymer. This difference notwithstanding, all side-chain and alphaN protecting groups, as well as coupling chemistries, employed in solid-phase synthesis have been successfully applied in solution-phase synthesis, and vice-versa, with few exceptions.
During the 1970s several groups were actively developing milder methods for SPPS that avoided the use of liquid HF as for the final deprotection/cleavage reagent. While a variety of milder graduated acid lability systems were devised, the method that rose to general applicability was the orthogonal system of Fmoc/tBu chemistry. This strategy – developed by R.C. Sheppard at Cambridge University – differs from Boc/Benzyl chemistry in that the side-chain and alphaN protecting groups are removed under conditions that leave the other class entirely intact. In Fmoc/tBu chemistry, a mild base – usually piperidine (pKa = 11.1) – is employed for iterative N 9-fluorenylmethoxycarbonyl (Fmoc) deprotection, while global side-chain deprotection/cleavage is accomplished with TFA (Scheme 2).
It must be emphasized that the more traditional Boc/Benzyl and Fmoc/tBu chemistries, while differing in chemical minutiae, are fundamentally the same process. In both cases, the target peptide chain is assembled in a stepwise fashion from alphaN- and side-chain protected amino acids. In both cases, the ‘transient’ alphaN amino protection is employed solely during chain elongation (the coupling reaction) and then removed for the subsequent coupling reaction. Lastly, in both cases, the final step – global side-chain deprotection and cleavage of the peptide-resin anchorage – is accomplished by acidolysis, and the target peptide isolated by trituration from ether and purified by reversed-phase high performance liquid chromatography (RP-HPLC).
.....
Download the full 65 page guide here:
About CSBio: For over 30 years, CSBio, a leading peptide and peptide synthesizer manufacturing company located in Silicon Valley, California, has been providing cGMP peptides and automated peptide synthesizers to the global pharmaceutical community. CSBio’s peptide products and peptide synthesizers can be found in production laboratories, universities, and pharmaceutical companies worldwide.
Want to know when new
Resources are released? Be notified
Related Posts
Synthesizing a 132-mer peptide with high purity
Synthesis of peptides at any length requires extreme attention to detail for accuracy and purity of the desired compound...
Research Scale Peptide Synthesizer Comparison - A Guide to Choosing a System that Fits Your Needs
A guide for choosing a research scale peptide synthesizer, comparing the most popular synthesizers on the market today.
About Us
CSBio is a leading peptide instrumentation manufacturing company located in Silicon Valley, California.
CSBio provides research scale peptide synthesizers, pilot scale peptide synthesizers, commerical scale peptide synthesizers, and DNA/RNA oligonucleotide synthesizers.